



July 10, 2024
This is the fourth in our series of regulatory updates on the EU AI Act.
Emergo by UL has been reporting on the landmark legislation that is the Artificial Intelligence Act (AIA). As the AIA was given the green light by EU institutions and the finish line is in sight, it seems fitting to conclude our regulatory series with a brief recap, as well as a focus on the high-risk AI systems on which the regulation is mainly concerned.
Recap
The EU AIA introduces a risk-based classification for AI systems. Medical devices employing AI (and ML) are likely to be classified as high-risk AI systems. This categorization imposes certain requirements based on AI best practices. Non-compliance with these requirements and indeed with the regulation can result in hefty fines being levied on medical device manufacturers.
For high-risk AI systems already on the market, the AIA shall only apply before the end of the stated transition period if there will be significant changes to the design or intended purpose.
The AIA imposes some additional requirements on economic operators (and operators) in the supply chain.
EU AI office is now open
Since our last regulatory update, the EU AI office has become operational. The AI office plays a key role in enforcement and oversight and sits within the EU Commission (EC). Amongst other tasks, it will enable the implementation of the AIA by supporting Member State governance bodies, enforce rules for GPAI models and promote collaboration amongst key stakeholders in the AI ecosystem. This will help foster a strategic approach to AI development that is innovative while also being safe and ethical in order to assure AI-enabled devices placed on the EU market are trustworthy.
Definitions
Definitions introduced by the AIA that are of relevance to this update have been extracted verbatim from the AIA:
| AI office | the EC’s function of contributing to the implementation, monitoring and supervision of AI systems and general-purpose AI models, and AI governance, is provided for in the Commission Decision of 24 January 2024 |
| Safety component | a component of a product or of an AI system that fulfills a safety function for that product or AI system, or the failure or malfunctioning of which endangers the health and safety of persons or property |
| *Provider | a natural or legal person, public authority, agency or other body that develops an AI system or a general-purpose AI model or that has an AI system or a general-purpose AI model developed and places it on the market or puts the AI system into service under its own name or trademark, whether for payment or free of charge |
*Based on contextual usage, legal manufacturers of AI-enabled medical devices may also be considered providers.
Classification as high-risk
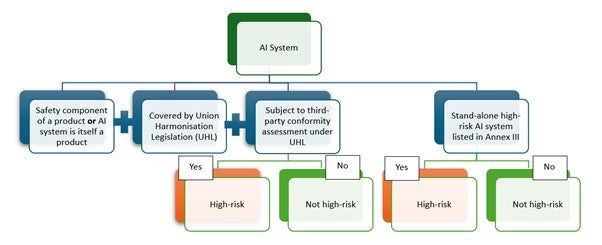
It should be noted that while a considerable number of AI-/ML-enabled medical devices are likely to fall under the high-risk category, there may be exceptions. Manufacturers should perform and document a risk classification based on the criteria outlined in the legislation (Figure 1).
With regards to the interplay between the AIA and the EU MDR/IVDR, the classification of an AI-/ML-enabled device as high-risk under the AIA does not automatically translate to a high-risk classification under EU MDR/IVDR and vice versa. The two classifications must be reviewed, assessed and evaluated separately.
Figure 1: Classification of AI system as High-risk under the EU AIA
Requirements for providers of high-risk AI systems
Before placement on the market or putting into service, providers of high-risk AI systems have several obligations to fulfill. Key requirements are outlined in Figure 2.
Where a provider believes that an AI system referred to in Annex III does not pose a significant risk of harm to the health, safety, or fundamental rights of natural persons, it is not high-risk. In this situation, the provider shall document an assessment before placing the AI system on the market or putting it into service. The documented assessment shall be made available to competent authorities upon request, and the provider must still register the AI system in the EU database.
With regards to registration in the EU AI database, which is separate from EUDAMED, it was reported in the MDCG New Technologies working group meeting, at the end of May (28 and 29) that to avoid double registrations, manufacturers of AI-enabled medical devices will not need to be registered under the database to be established under the AIA, as EUDAMED registration will take precedence. This is to be confirmed.
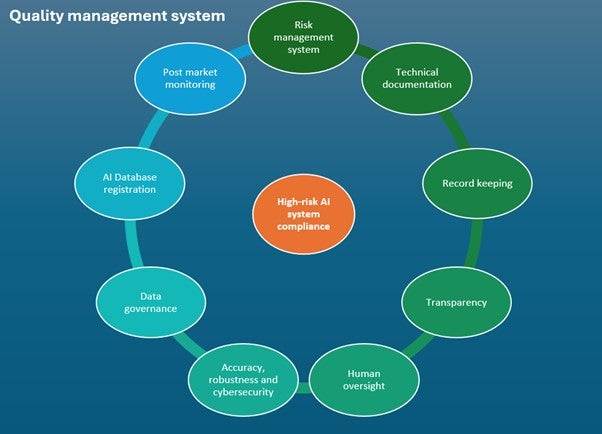
Figure 2: Requirements for compliance of high-risk AI systems under the AIA
Commonalities with EU MDR/IVDR
Although the AIA represents another regulatory framework that manufacturers of applicable devices must comply with, as both are New Legislative Framework legislation, there are commonalities between the two frameworks that means that medical device manufacturers are better placed than others that fall under the AIA to achieve compliance. These include:
- Quality management system (QMS).
- Conformity assessment.
- Technical documentation.
- Risk management system (including cybersecurity).
- Documentation and record keeping.
- Appointment of an EU AR (where applicable).
- Declaration of conformity.
- CE marking.
- Cooperation with competent authorities; including performing corrective actions and information sharing.
Additional regulatory burden
There are of course additional regulatory requirements that providers of AI-enabled medical devices must comply with. These include:
- Data and data governance; assuring high-quality datasets for testing, training and validating. It should be noted that where necessary for conformity assessment Notified Bodies are required to have access to this data.
- Automatic logging of events to assure traceability.
- Human oversight; to ensure the safe, ethical and responsible use of AI systems.
- Transparency; including mandatory information to be provided to users.
- Accessibility requirements; to foster equality for all users and prevent bias.
- Registration of devices in the EU AI database.
Additionally, although the AIA and MDR share the requirement for a QMS in common. An ISO/IEC 42001 Artificial Intelligent Management System will likely be expected to fulfil specific requirements. This may be integrated into an existing ISO 13485 QMS.
The same approach applies to risk management, where manufacturers will potentially need to consider ISO/IEC 23894 for AI-specific risks, in addition to ISO 14971.
Route to market
AI-enabled medical devices that require Notified Body involvement as part of conformity assessment under the EU MDR or IVDR will undergo conformity assessment under the AIA at the same time; the plan is to add additional product codes that cover AIA compliance to Notified Bodies already certified under the EU MDR or IVDR.
Certain high-risk systems under Annex III and devices that do not require Notified Body involvement under the EU MDR or IVDR will demonstrate conformity via internal control i.e. self-declaration.

Figure 3: High-level summary outlining the route to market for a high-risk AI system under the EU AIA
*EUDAMED registration may fulfill the requirements for database registration. This is to be confirmed.
Concluding remarks
The AIA intends to put in place a framework that complements legislation such as the EU MDR and does not result in duplicate processes or documentation. The recommendation is to have a single set of documentation where practical. A single declaration of conformity and single CE marking will also be expected.
The AIA requires medical device manufacturers to comply with additional requirements; this may for example, require consideration of additional standards, or implementing requirements cumulatively to account for any differences. Manufacturers should perform a gap analysis of the requirements and integrate where possible.
As the AIA enters into force we expect to see published supporting guidance documents. Only time and experience will tell how this landmark legislation will be implemented in practice, as well as what the full impact on the medical device industry will be.




X
Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…
Characters remaining