



May 10, 2024
By Sade Sobande
This is the first in a series of regulatory updates on the EU Artificial Intelligence Act.
On March 13, the European (EU) Artificial Intelligence Act (AIA) was adopted by the European Parliament. This marked a significant milestone in a journey that officially began on April 21, 2021 when the Act was first proposed by the EU Commission.
The adoption of the AIA means that the world now has a comprehensive framework for regulation of AI-enabled devices. As with the EU General Data Protection Regulation, it is envisaged that there may be a “Brussels Effect” and the AIA will potentially become the global standard that subsequent AI frameworks are based on.
In this first of our series on the AIA, Emergo by UL covers the key takeaways related to scope, classification and conformity assessment that manufacturers will need to be aware of. We continue to monitor the effect of the AIA on the medical device industry.
The AIA Is horizontal legislation
The AIA applies across all industry sectors and is considered part of the New Legislative Framework (NLF) portfolio of legislation. As a regulation, it is also applicable across all EU Member states. For medical device manufacturers, this means that if a medical device incorporates AI or indeed ML-enabled device functions, then the AIA will be applicable to those functionalities. The AIA also prescribes conditions when compliance of the AI product requires the CE marking affixed as a demonstration the products are compliant.
Scope of the AIA
Providers and deployers of AI systems and/or general-purpose AI (GPAI) models who intend to make available or put into service these technologies on the EU market fall in scope of the AIA. This is irrespective of whether or not the providers and deployers are established in the EU, so long as the output (at a minimum) is intended to be used within the EU.
The definitions of AI system and GPAI are:
‘AI system’ means a machine-based system that is designed to operate with varying levels of autonomy and that may exhibit adaptiveness after deployment, and that, for explicit or implicit objectives, infers, from the input it receives, how to generate outputs such as predictions, content, recommendations, or decisions that can influence physical or virtual environments.
‘general-purpose AI model’ means an AI model, including where such an AI model is trained with a large amount of data using self-supervision at scale, that displays significant generality and is capable of competently performing a wide range of distinct tasks regardless of the way the model is placed on the market and that can be integrated into a variety of downstream systems or applications, except AI models that are used for research, development or prototyping activities before they are placed on the market.
AI systems placed on the market, put into service or used by public and private entities for military, defense or national security purposes, are out of scope of the regulation.
AI systems and models, including their output, intended to be used for the sole purpose of scientific research and development are also out of scope.
Classification in the AIA
A risk-based approach is taken to classify AI systems and GPAI models. AI systems are classified according to four categories (Figure 1). Medical devices including IVDs that incorporate AI/ML-enabled device functions will likely be classified as high-risk AI systems. The requirements for high-risk AI systems include: (1) Data governance; (2) Quality Management System (QMS); (3) Technical documentation; (4) Record keeping; (5) Transparency; (6) Human oversight; (7) Accuracy, robustness and cybersecurity; (8) Conformity assessment based on either internal controls (self-declaration) or Notified Body involvement.
While some of these requirements seemingly overlap with the requirements under the EU Medical Devices Regulaton (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR), the devil is in the detail, and manufacturers will need to conduct a gap analysis in order to remediate any process shortcomings. Additionally, due to the specific requirements under each regulation, it may be more coherent to have, for example, distinct technical documentation to support compliance under the AIA, MDR and/or IVDR.
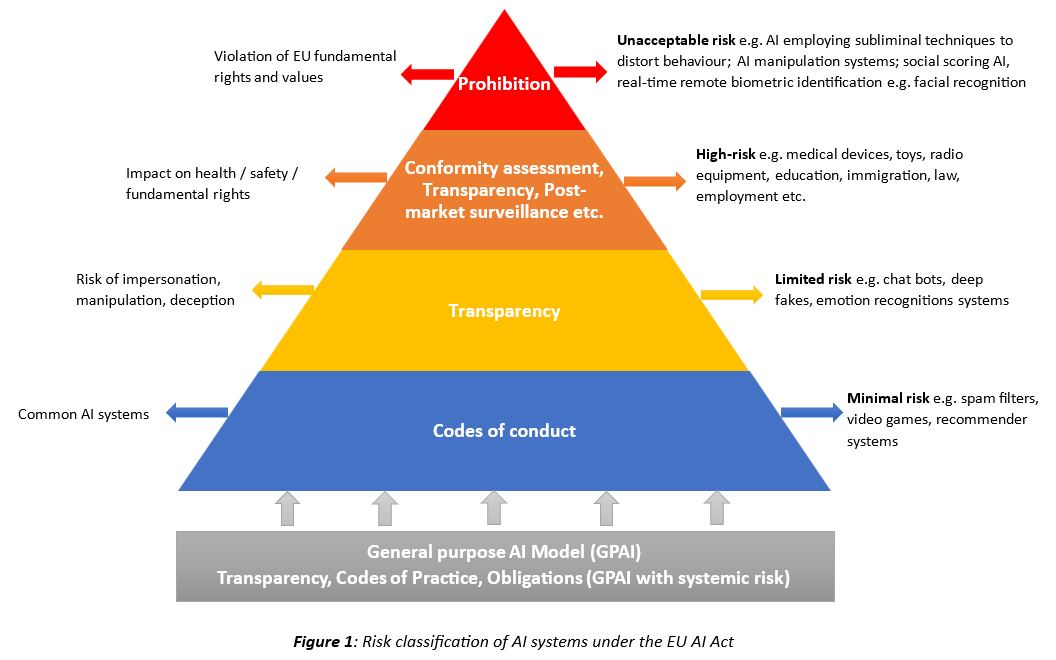
With regards to GPAI models, special risk classification rules apply; they are classified as GPAI models and GPAI models with systemic risks, to which more stringent obligations apply. GPAI models will need to comply with transparency obligations and codes of practice which may be used as presumption of conformity with the AIA. Additionally, for GPAI models with systemic risk, there are the following requirements: (1) Notification to the Commission; (2) Perform model evaluation; (3) Assess and mitigate systemic risks (4) Vigilance reporting to AI office and national competent authorities; (4) Cybersecurity protection.
GPAI models that are integrated into AI systems or form part of the AI system are considered GPAI systems and shall go through the AI system classification process. A GPAI model presenting systemic risks that is then integrated into a high-risk AI system will therefore have to comply with requirements for high-risk AI systems, as well as requirements for GPAI models with systemic risk for the AI model part.
AI systems posing an unacceptable risk due to violation of EU fundamental rights and values are explicitly prohibited.
Conformity Assessment in the AIA
Conformity Assessment is not new to the medical device and IVD industries, so here there will be some overlap. For high-risk AI systems conformity is either via internal control (i.e. self-declaration) or Notified Body involvement. With respect to medical devices and IVDs, where conformity assessment under the MDR and IVDR already requires Notified Body involvement, the intention is for conformity assessment under the AIA to be done at the same time. The practical implications of this will become clear as this starts to be implemented, but the planned strategy is to add product codes for the AIA to the scope of those Notified Bodies that intended to implement it.
Operator Obligations
The AIA delineates obligations for five main “operators” (similar to “economic operators” under the NLF legislation such as the MDR and IVDR): providers, deployers, authorized representatives, importers and distributors. Each of these operators has specific roles to play within the AI value chain. Some of the key roles of each are outlined in Figure 2.
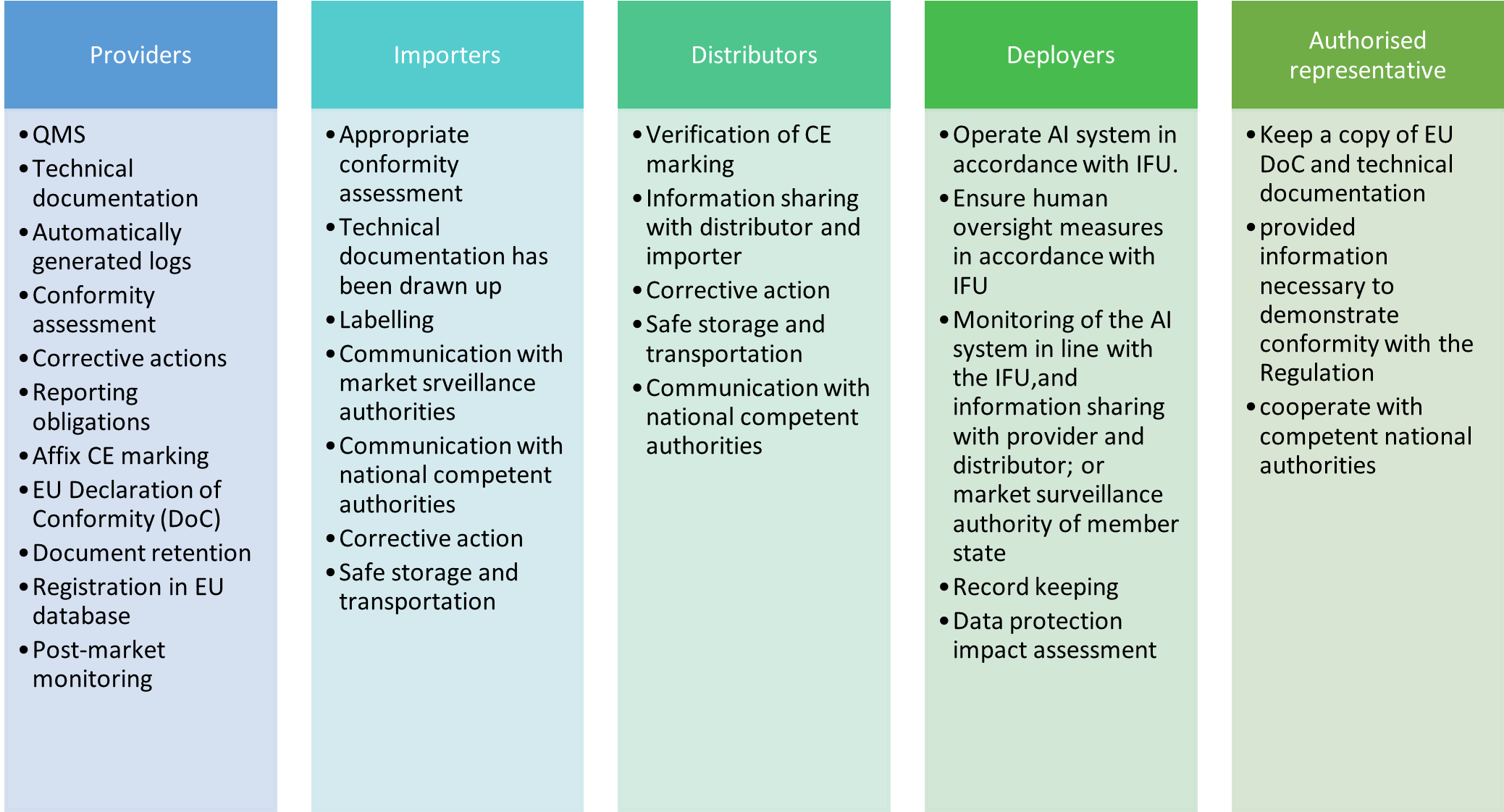
Distributors, importers, deployers or third-parties may be considered a provider if they place on the market or put into service a high-risk AI system under their name or trademark, modify the intended purpose of the system, or make a substantial modification to the system.
Concluding Remarks
As with any new piece of legislation there will be challenges to overcome, and it is yet to be seen how the AIA will unfold in practice. However this is the time for organizations to put in place plans and strategies to comply with the AIA.
Sade Sobande is lead quality and regulatory affairs consultant at Emergo by UL.



Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.