



March 26, 2025
By Sarah Fitzgerald
Importance of reviewing warning letters
By reviewing relevant U.S. Food and Drug Administration (FDA) warning letters, manufacturers can better understand areas of frequent concern related to quality management systems (QMS) and regulatory affairs. This can allow companies to focus on areas of highest risk as they continually assess and improve their internal procedures and processes. It also helps a company better understand significant concerns from the FDA.
Warning letters in 2024
The FDA’s Center for Devices and Radiological Health (CDRH) issued 529 warning letters in 2024,[1] most related to drugs, tobacco products or food products. Of those, 8% (44)[2] were issued for medical devices. Most warning letters include multiple issues. Please see our previous blog on 2024 warning letters for additional details.
Categorization of warning letters and relevance to LDT Final Rule stages
The Laboratory Developed Test (LDT) Final Rule requires standard medical device requirements to be met over four years and includes five stages, each with its specific requirements.
In summary:
- Stage 1: May 6, 2025: Establish and follow procedures for complaint management, adverse event reporting, and corrections and removal.
- Stage 2: May 6, 2026: Establish and follow procedures for establishment registration, device listing, labeling and investigational device use.
- Stage 3: May 6, 2027: Establish a complete QMS compliant with FDA requirements (including ISO 13485:2016).
- Stage 4 and 5: November 6, 2027 and May 6, 2028: Submit appropriate premarket applications to the FDA (such as premarket applications [PMAs], de novo classification requests [de novos] and 510(k)s), as appropriate for the individual devices.
For further details, please see our previous blog on the LDT Final Rule.
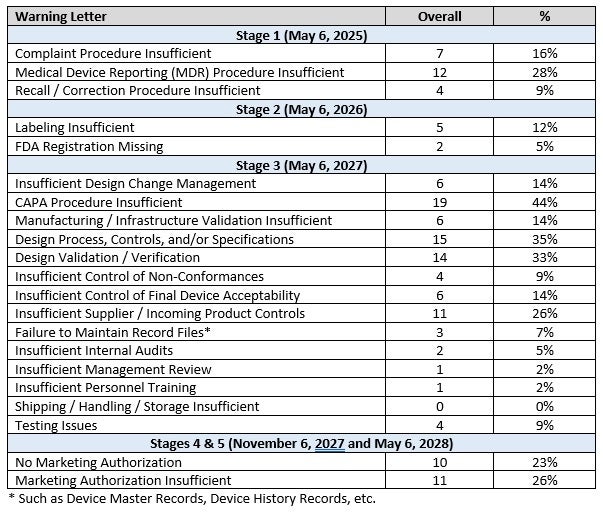
Table 1 provides a summary of all deficiencies and calculates a percentage of the total of the medical device-related warning letters. These have been organized to align with the LDT requirements stages.
Table 1. Warning letters and categories overview
Table 2 summarizes the most frequently cited categories for 2023 and 2024. The same categories are generally of the most concern for both years, although with the greater number of warning letters in 2024 there appears to be less concentration of issues.
Table 2. Most frequently cited categories in warning letters
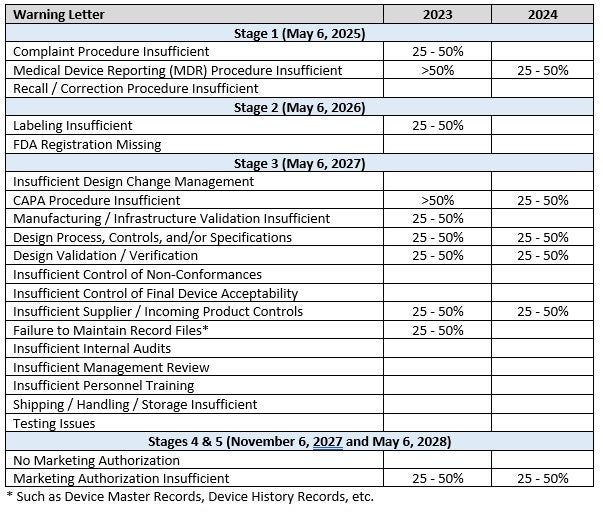
These tables illustrate that each stage of FDA requirements has critical issues that the FDA often finds are insufficient in companies. Compliance with each stage is important.
Timeline considerations
It is important to consider lead time for implementing FDA requirements. Implementing a few procedures, such as for Stage 1 and 2, may only take 1-4 months. However, this may not include resource considerations, including related to personnel. Therefore, we recommend that companies begin work on these requirements at the beginning of the relevant year or as soon as possible.
Implementing an entirely FDA-compliant QMS (Stage 3) generally is expected to take 12-18 months. Therefore, we recommend that companies start working on these no later than the beginning of 2026.
Submitting appropriate premarket submissions to the FDA (Stages 4 and 5) vary depending on how much information needs to be documented and what information is already available. Assuming any necessary clinical data is already available and does not need to be gathered, many companies find that compiling a premarket application (PMA) for a high-risk device often takes 12-24 months, a de novo is 9-12 months, and a 510(k) is 4-6 months.
Most LDTs are expected to require a 510(k) or de novo, but if the type of submission is unknown, the regulatory pathway/strategy should first be determined. Of note, it is often advisable to submit a pre-submission Q-submission (pre-subs) to the FDA before submitting a premarket submission in order to align on expectations, such as clinical evidence and other testing expectations. These meetings can help confirm that testing does not need to be redone and that the premarket submission itself will be authorized by the FDA. Therefore, we recommend that companies determine their regulatory pathway to market in 2025-2026 and consider pre-subs in 2026 to allow time to take action on any FDA feedback before submitting the premarket submission.
These are, of course, simply general timelines for many companies. The timeline may be affected by various factors and should be carefully considered by each company.
LDT manufacturers should pay careful attention to appropriately implementing the staged requirements of the FDA’s LDT Final Rule. Each stage includes requirements that the FDA has found frequently deficient in warning letters, as indicated in Tables 1 and 2.
We at Emergo by UL encourage LDT manufacturers to begin work on implementing the requirements, considering likely project timelines. We note that especially Stages 3, 4 and 5 can have long timelines and recommend seriously considering these requirements in 2025 to assemble a timeline based on individual company resources and factors. We are available to assist with medical device QMS and regulatory considerations, including for LDTs.
[1] Posted warning letters as of January 24, 2025.
[2] Note: The FDA CDRH annual report noted there were 44 warning letters issued, but a search of the FDA database on January 24. 2024 only located 43 as it may take a few months before posting on the FDA database. This report is based on the 43 that could be analyzed.



X
Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…