



May 31, 2024
A recent round of inspections by the Dutch Health and Youth Inspectorate (IGJ) has found that more than one-third of European Authorized Representatives (ARs) operating in the Netherlands have yet to meet all requirements applicable under the European Medical Devices Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR).
European Authorized Representatives under supervision
European ARs are the gateway to the European Union (EU) for non-EU based medical device and IVD manufacturers. As such it isn’t surprising that with the new Regulations, ARs must comply with stricter requirements, as well:
- ARs need to comply with legal requirements based on European legislation, such as the MDR and IVDR
- Competent Authorities (CAs) across the EU supervise ARs in the member states in which they’re based
- ARs are held legally liable for defective devices on the same basis as, and jointly and severally with, the manufacturers they represent
The Dutch Health and Youth Care Inspectorate (IGJ) supervises ARs based in the Netherlands, and recently published a report on the outcome of the their inspections of 24 ARs based in the country. (Emergo by UL’s AR business, Emergo Europe B.V., is based in the Netherlands and was included in the report.)
What was inspected?
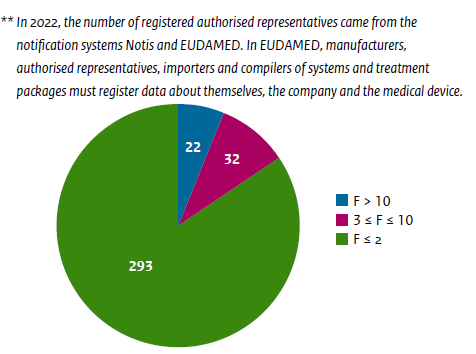
In the Netherlands alone, 347 ARs are registered in the Dutch Database, NOTIS. Picture 1 below shows the registered ARs and the number of devices they represent.
Requirements that need to be met by ARs are listed in Article 11, 12 and 15 of the MDR and IVDR. Additionally, guidance documents are published by the Medical Devices Coordination Group (MDCG), such as MDCG 2022-16 Guidance on Authorised Representatives Regulation (EU) 2017/745 and Regulation (EU) 2017/746. Although not law, these MDCG documents are expected to be taken into account by those that are addressed in these MDCG documents.
The IGJ is only allowed to act based on law, and as such the supervision framework for ARs is based on the MDR and IVDR. Nevertheless, recommendations for improvement have also been given, most of these coming from MDCG 2022-16.
Main inspection topics include:
- The mandate between the AR and the manufacturer (Article 11(2,3,4,6) MDR/IVDR)
- The AR meets the basic requirements which follow from Article 11 and 12 MDR/IVDR in respect of the manufacturers they represent and in respect of the medical devices that are placed onto the European market by those manufacturers
- The AR has appointed one or more Persons Responsible for Regulatory Compliance (PRRC) (Article 15 MDR/IVDR)
The IGJ inspection outcomes
More than one-third of the ARs in the IGJ’s inspection report do not meet the basic requirements that follow from the MDR and IVDR. The IGJ additionally emphasized it is important that ARs also meet the guidelines established in MDCG 2022-16, even though it is not a legally binding document. The IGJ also listed several points for improvement.
Recommended criteria for selecting a European AR
So, in light of the IGJ findings, what are basic requirements to pay attention to when you are searching for a (new) AR?
- The AR must be a physical address in the EU, not just a postbox address. Several ARs in the IGJ report did not appear to have a physical address in the Netherlands where people are doing the actual work for the AR service. This is not acceptable, as CAs would in these cases not be able to get in touch with the AR of a manufacturer in the event of a vigilance case, for example.
- There must be a mandate in place between the AR and the manufacturer in accordance with Article 11(3) MDR/IVDR. This was the situation in all cases; however, in some cases the mandate met the requirements discussed in this article.
- The mandate should clearly list the device families that the AR is representing for the manufacturer. This was not always the case in the IGJ report.
- If the manufacturer and AR are part of the same organization, there still must be a mandate in accordance with Article 11(3) MDR/IVDR. The manufacturer is not allowed to delegate any of the tasks listed in Article 10 MDR/IVDR to the AR. Both the manufacturer and the AR need to have liability insurance (Article 11(5) MDR/IVDR. And both need to have their own PRRC who meets the requirements of Article 15 MDR/IVDR.
- Your AR must have verified that your Technical Documentation and EU Declaration of Conformity have been drawn up, and, you have a valid CE certificate in place (if applicable). So don’t be suspicious if this is asked for by your AR, be suspicious if they don’t ask for it!
Some of the opportunities for improvement were given based on best practice. For example, “A number of authorized representatives were able to thoroughly demonstrate the added value of their PRRCs, for example, due to the fact that the PRRC was involved in the exchange of data with manufacturers and in the verification of documents required by law.”
Conclusions and takeaways
When searching for a new AR, make sure your potential in-country representative meets the basic requirements laid out in Article 11, 12 and 15 MDR/IVDR. The IGJ report gave many ARs opportunities for improvement, most of them related to guidelines in MDCG 2022-16. Not all ARs follow this guidance, or suggestions for improvement which followed from the report. This does not imply they aren’t compliant with the basic requirements of the MDR/IVDR, but they may be less capable of assisting manufacturers with valuable market insights and regulatory support when needed. Emergo by UL is well-versed in MDCG guidances and EU Regulations, and takes its role as gatekeeper to the European market seriously. We will keep you informed of further EU regulatory updates as they occur.




